

COPD是一种以持续性、进行性的气流受限为特征的慢性气道炎症疾病。中国成人肺部健康研究调查显示,我国40岁及以上人群COPD患病率达13.7%,总患病人数近1亿,已成为严重影响我国居民健康的主要慢性疾病。COPD是由外界环境因素和自身遗传因素共同作用的多因素异质性疾病,外界环境因素如香烟烟雾、大气污染物等与COPD的发生、发展密切相关。随着我国城市化进程的加快,空气质量控制形势严峻,大气污染对人群健康的影响备受社会关注。国内外多个环境流行病学研究发现,大气污染参与了COPD的发生、发展,其不仅是COPD的重要危险因素,还是引起COPD急性发作、住院率及病死率增加的主要非感染因素。大气污染物对COPD影响的具体分子机制目前尚不明确,作为连接环境因素与遗传因素的"桥梁" ,表观遗传学相关的研究越来越多,从表观遗传学的角度出发可以更好地阐释大气污染物对COPD影响的可能机制。
表观遗传学是在不改变DNA序列的情况下通过DNA修饰作用改变基因表达的一种可遗传性变化,主要包括DNA甲基化、组蛋白修饰、非编码RNA修饰等。表观遗传学将环境与遗传因素紧密结合在一起,随着研究深入,已发现表观遗传学的改变从根本上是由环境因素作用的结果。环境中的化学、物理、生物及精神心理和营养饮食等因素均可导致表观遗传学改变,其中的某些变化或可成为环境暴露的潜在生物学标志物。现已发现,大气污染物作为重要的环境暴露因子,可引起机体内出现多种表观遗传学改变。
1、DNA甲基化
DNA甲基化是目前研究最为深入的表观遗传学修饰,在DNA甲基转移酶(DNA methyltransferase,DNMT)催化下完成。甲基化可引起DNA构像、染色质结构、DNA与蛋白质作用方式等发生改变,DNA高甲基化通常抑制基因表达,而去甲基化则促进基因的转录和表达。大气污染物可通过改变DNA甲基化水平从而参与COPD的发生、发展,DNA不同位点的甲基化或可作为大气污染暴露对机体健康影响的潜在生物标志物。
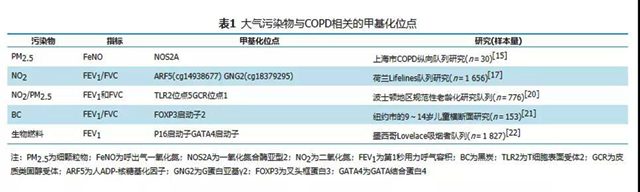
气道炎症是COPD重要的病理生理改变,呼出气一氧化氮(fraction of exhaled nitric oxide,FeNO)被认为是一种敏感的气道炎症标志物,可较好的反映患者气道内炎症状态,经常被用于评估与大气污染暴露相关的不良呼吸影响。FeNO由一氧化氮合酶亚型2(nitric oxide synthase 2A gene,NOS2A)参与调控生成。既往研究发现细颗粒物(fine particulate matter,PM2.5)暴露与FeNO水平相关,但不明确PM2.5是如何引发急性气道炎症的。Chen等对上海市某城区30例COPD患者进行了6次短期重复性健康调查,发现PM2.5暴露与口腔黏膜组织内NOS2A启动子区甲基化的减少和FeNO的增加显著相关,表明PM2.5可能通过改变NOS2A启动子区的甲基化状态而调控一氧化氮(nitric oxide,NO)的产生,进一步导致气道组织损伤、加重气道内炎症反应。诱导型一氧化氮合酶(inducible nitric oxide synthase,iNOS)是由NOS2A编码表达的调控气道中FeNO生成的一种关键限速酶,郑鑫等通过动物实验进一步证实了PM2.5可降低iNOS基因DNA启动子区甲基化水平,促进iNOS基因表达和转录,促进NO生成增加。这些研究可更好地将大气污染物与COPD的不良健康结局的机制联系起来。
二氧化氮(nitrogen dioxide,NO2)是影响COPD发生、发展的主要气态污染物。荷兰LifeLines大型队列研究发现NO2暴露与人群FVC和第1秒用力呼气容积(forced expiratory volume in one second,FEV1)/FVC下降相关,并与多种DNA位点异常甲基化相关,为探究NO2是否通过改变DNA甲基化水平引起肺功能受损,进一步进行中介分析效应分析发现与NO2暴露相关的CPG位点中,cg14938677介导了NO2暴露与FVC的关系,cg14938677和cg18379295介导了NO2暴露与FEV1/FVC的关系,这些位点的甲基化被认为是NO2暴露与肺功能相关的潜在生物标志物。另一项韩国的COPD人群队列研究也得到了类似结果,提示NO2通过改变DNA甲基化水平影响肺功能。为探究NO2对COPD表型的影响,Zhang等将小鼠暴露于NO2环境下构建COPD的肺气肿表型模型,并设置DNMT抑制剂+NO2为对照组。研究发现与对照组相比,单纯暴露于NO2组的小鼠FEV1/FVC及缺氧程度更重,即DNMT抑制剂可有效改善肺气肿的形成,或具有潜在治疗作用。其他几个已发现大气污染物与COPD相关的甲基化位点见表1。
2、组蛋白修饰
组蛋白修饰是另一种重要的表观遗传修饰。组蛋白是真核生物细胞中与DNA碱基对共同组成核小体单元的碱性蛋白质,其末端氨基酸残基可发生多种共价修饰,包括乙酰化/去乙酰化、甲基化、磷酸化等。其中,乙酰化/去乙酰化是最常见、研究最多的组蛋白修饰。组蛋白在乙酰转移酶(histone acetyltransferase,HAT)催化作用下发生乙酰化,其表面的负电荷被乙酰基中和,与DNA碱基的结合能力减弱,核小体结构松弛,使DNA与相关调控元件如转录因子、促转录因子结合并启动转录,最终上调基因表达。与之相反,组蛋白去乙酰化则是在脱乙酰转移酶(histone deacetylase,HDAC)催化下组蛋白末端乙酰基被去除,通常组蛋白去乙酰化后基因的转录和表达处于抑制状态。因此,HAT和HDAC对基因的转录和/或沉默起关键性调节作用。
慢性炎症反应是COPD重要的发病机制,如细胞因子、趋化因子、炎性蛋白酶等,这些因子的产生增加受促炎转录因子如核因子κB(nuclear factor kappa-B,NF-κB)的调节,而HDAC活性下降和/或HAT增高均可激活NF-κB,导致促炎基因表达增加,其中HDAC2活性下降是诱导炎性基因表达的关键因素。既往研究发现COPD患者的肺组织、肺泡巨噬细胞、外周血和肌肉组织中HDAC2表达下降,导致组蛋白去乙酰化作用减弱,NF-κB激活,进而启动一系列炎症反应。环氧化酶2(cyclooxygenase-2,COX-2)亦是炎症反应过程中的关键促炎诱导因子,与多种炎性介质共同参与COPD的气道炎症反应,柴油机尾气颗粒物可通过降低HDAC1的活性、促进COX-2基因启动子H4组蛋白乙酰化,导致COX-2转录增多,促进炎症反应。颗粒物是我国各城市大气环境的主要污染物,PM10可引起HAT活性升高、IL-8基因启动子区域的组蛋白H4乙酰化水平升高,IL-8产生增多,引起肺部持续的炎症反应。综上,大气污染物可通过降低HDAC1的活性和/或提高HAT的活性引起COPD人群组蛋白乙酰化水平升高,促进促炎基因的转录,引发肺内持续的炎症反应,未来可对HAT和/或DHAC酶活性角度开展深入研究,为COPD治疗提供科学参考。
3、miRNA
miRNA是一类长度为20~24个核苷酸的非编码单链小分子RNA,通过至少6~8 nt长的互补序列与靶标基因的3′端非编码序列相结合,抑制靶向基因mRNA的翻译或介导其降解,从基因转录后水平调控靶基因的表达。近年研究发现miRNA在环境化学物质引起的毒理学过程中起着十分重要的作用,发现污染物可通过改变某些miRNA表达,对机体产生致病效应,并陆续开展了相关机制研究(表2)。

4、LncRNA
LncRNA亦属于非编码RNA,其转录本长度大于200 nt,一般不编码蛋白质,可在表观遗传水平、转录水平、转录后水平调节基因的表达。生物体内许多信号转导通路都受LncRNA的调控,在细胞分化、增殖和凋亡等过程中具有重要作用。
目前LncRNA主要功能和参与COPD的病理生理机制仍不清楚,关于大气污染相关的LncRNA与COPD的研究亦较少,且多为细胞实验。PM2.5可引起人支气管上皮细胞多种LncRNA异常表达改变,部分LncRNA与PM2.5诱导的细胞毒性和基因毒性有关。LncRNAMEG3可靶向调节细胞凋亡以及自噬相关蛋白,Li等分析了暴露于交通相关PM2.5下的人支气管上皮细胞内LncRNA表达谱,共有1 292条LncRNA上调,1 362条LncRNA上调,通过siRNA干扰表达明显异常的LncRNAMEG3后,p53及凋亡蛋白caspase-3表达下调,细胞凋亡率减低,提示LncRNAMEG3可能参与PM2.5介导的细胞凋亡和自噬。Xu等发现,PM2.5可引起人支气管上皮细胞细胞周期停滞,经PM2.5处理后,LncRNA LINC00341在人支气管上皮细胞中显著上调,通过抑制LncRNA LINC00341的表达可逆转PM2.5诱导的p21表达和G2/M期细胞周期阻滞。上述结果表明,LncRNALINC00341可能通过调节p21的表达来调节PM2.5诱导的细胞周期阻滞,从而介导PM2.5对支气管上皮细胞的毒性作用。尚需明确调控大气污染物对支气管上皮细胞毒性作用的关键LncRNA分子。
5、展望
表观遗传学受环境因素的影响并可反映环境的变化,在环境、基因表型与疾病易感性之间起关键作用,提供了可靠的生物学机制解释环境暴露与疾病之间的关系的生物学机制。COPD是由环境和基因共同作用的多因素复杂疾病,深入研究表观遗传学有助于深入了解大气污染对COPD发生、发展的影响。目前关于表观遗传模式及各调控机制间的相互关系仍不明确,仍需在流行病学、细胞生物学、表观遗传学等方面开展研究,识别关键的表观遗传调控机制。
参考文献:段瑞瑞, 牛宏涛, 于涛,等. 大气污染对慢性阻塞性肺疾病表观遗传学影响的研究进展[J]. 国际呼吸杂志, 2020, 40(14):6.



